Modelos Animais em Pesquisa Biomédica - Replicação de Artigo Científico
R
Data Science
Ciências Biológicas
Biomedicina
Danio rerio
CRISPR
Projeto de replicação de replicação de experimento baseado no artigo https://doi.org/10.1016/j.jbior.2022.100945 em que usaremos um desfecho e uma variável para replicar o estudo com modificações de metdologia que fazer-se-ão necessárias na implementação da pesquisa. Para o experimento foi seleciado o desfecho comparativo de KO e controle de mutação do gene enthd2
Authors
Affiliation
Lincolm Ferreira de Aguiar
Universidade Federal do Rio Grande do Sul - Biomedicina - Modelos Animais em Pesquisa Biomédica
Gabriel Morales Chiapin
Universidade Federal do Rio Grande do Sul - Biomedicina - Modelos Animais em Pesquisa Biomédica
Published
December 3, 2025
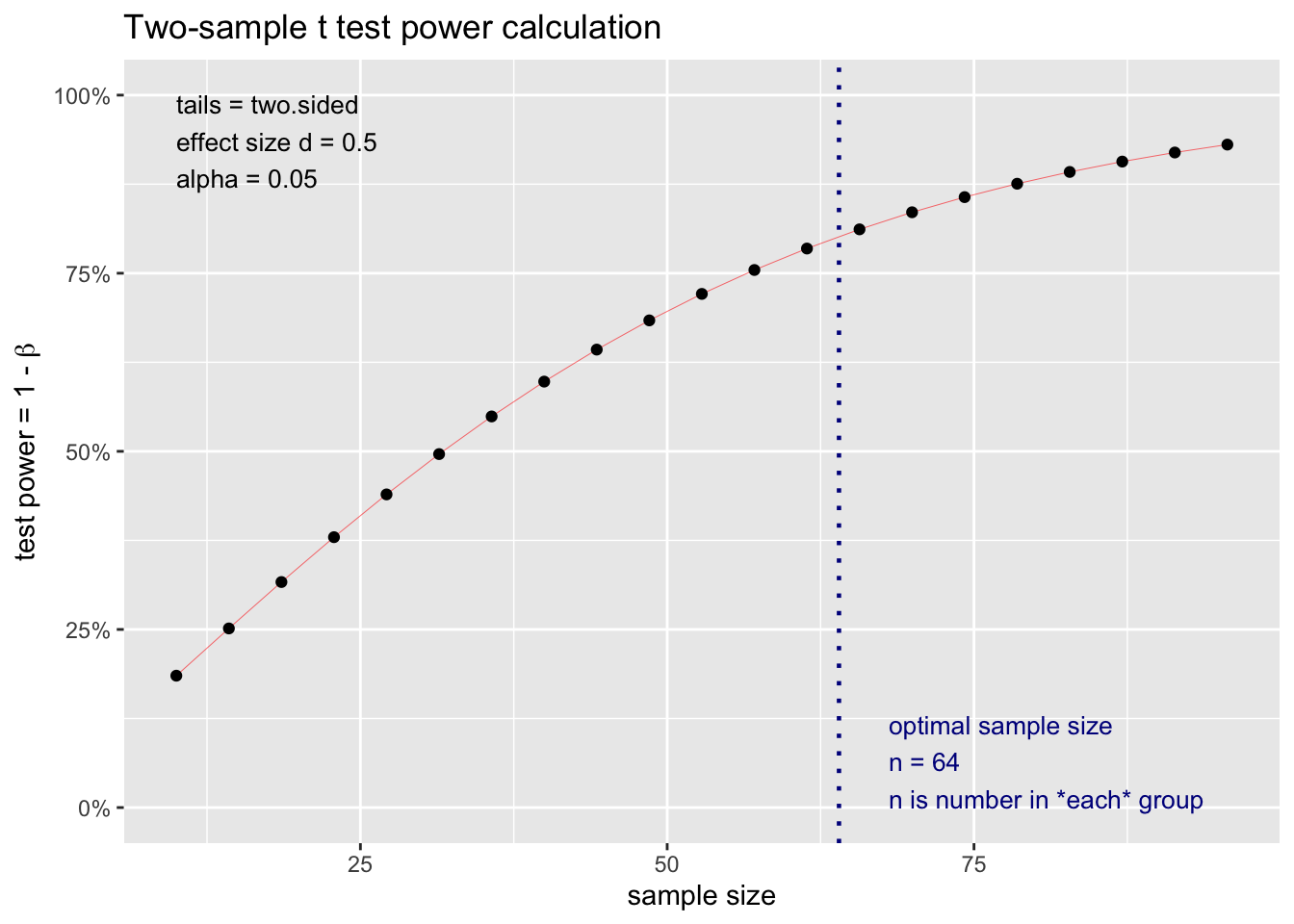
poder_estatistico <-0.8# 1-β, valor estimado para o estudo. nivel_confianca <-0.05# um nível de confianca de 0.05 (5%)numero_grupos <-2# o grupo KO se subdivide em: Classe 1, Classe 2 e Classe 3 quanto á má formação por CRISPRcohen_f <-0.5# O f de Cohen é o quão grande são as medidas de dispersão entre as médias# pois temos 2 pools (k=2) em relaçao a variabilidade dentro dos grupos# Sobre o uso de pool.# Com 64 animais, nõo precisamos de pool e podemos usar o equipamento QuantStudio da Applied Biosystems# disponível no laboratório Multi-usuário do ICBS, com acesso por agendamento.# Com 64 por grupo, podemos ter extrações de RNA relativamente baixas, mas com o numero de animais # é mais imporante a identidade (variabilidade) de animal por experimento. A sensibilidade estimada do # QuantStudio é de 1 a 10 cópias de DNA. # Optamos, por hora, em usar pool de animais.# https://www.ufrgs.br/agendamentoderecursos/Web/dashboard.php# Test t para o cálculo amostra porque temos 2 groups (KO e WT)d <-pwr.t.test(d = cohen_f, sig.level = nivel_confianca, power = poder_estatistico, type ="two.sample", alternative ="two.sided")plot(d)
# Número de animais que usaremos no estudo. Onde é n = 64 (do cálculo do tamanho amostral) x 2 gruposprj_n <-64*2prj_groups <-c("KO", "WT")
# fixamos o 2025 para ter reprodutibilidade. Em qualquer lugar que esse script rodar, # irá produzir a mesma sequencia de amostra.set.seed(2025)# Vamos alocar cada ovo de Danio rerio em de forma randomizada entre os grupos KO e WT. # Para isso, criamos uma sequenciaembryos <-paste0("embryo_", 1:prj_n) # E vamos alocar os animais nos grupos pela sequencia descrita nesse algoritmo.# Vamos separar entre WT e KO com (prj_n / 2) cadagroup_WT <-sample(embryos, size = prj_n /2)# O grupo KO receberá os embrioes de embryos que não estão alocados no grupo WTgroup_KO <-setdiff(embryos, group_WT)# Vamos transformar isso em uma forma apresentável (via uma data.frame)# KO=U# WT=G# A randomização aqui não tem efeito de cegamento, apenas alocação não enviesada de ovos por gruposgroups <-data.frame(embryo = embryos,group =ifelse(embryos %in% group_WT, "G", "U"))# E imprimimos o data.framegroups
embryo group
1 embryo_1 U
2 embryo_2 U
3 embryo_3 U
4 embryo_4 G
5 embryo_5 G
6 embryo_6 U
7 embryo_7 U
8 embryo_8 U
9 embryo_9 U
10 embryo_10 G
11 embryo_11 G
12 embryo_12 U
13 embryo_13 G
14 embryo_14 G
15 embryo_15 G
16 embryo_16 G
17 embryo_17 G
18 embryo_18 U
19 embryo_19 U
20 embryo_20 G
21 embryo_21 U
22 embryo_22 U
23 embryo_23 G
24 embryo_24 G
25 embryo_25 G
26 embryo_26 G
27 embryo_27 G
28 embryo_28 U
29 embryo_29 G
30 embryo_30 U
31 embryo_31 G
32 embryo_32 G
33 embryo_33 G
34 embryo_34 U
35 embryo_35 U
36 embryo_36 G
37 embryo_37 G
38 embryo_38 G
39 embryo_39 U
40 embryo_40 U
41 embryo_41 G
42 embryo_42 U
43 embryo_43 U
44 embryo_44 G
45 embryo_45 U
46 embryo_46 G
47 embryo_47 U
48 embryo_48 G
49 embryo_49 G
50 embryo_50 G
51 embryo_51 G
52 embryo_52 G
53 embryo_53 U
54 embryo_54 U
55 embryo_55 U
56 embryo_56 G
57 embryo_57 G
58 embryo_58 G
59 embryo_59 G
60 embryo_60 U
61 embryo_61 G
62 embryo_62 U
63 embryo_63 G
64 embryo_64 U
65 embryo_65 G
66 embryo_66 U
67 embryo_67 G
68 embryo_68 G
69 embryo_69 G
70 embryo_70 U
71 embryo_71 U
72 embryo_72 U
73 embryo_73 U
74 embryo_74 G
75 embryo_75 U
76 embryo_76 G
77 embryo_77 G
78 embryo_78 U
79 embryo_79 U
80 embryo_80 U
81 embryo_81 U
82 embryo_82 G
83 embryo_83 U
84 embryo_84 U
85 embryo_85 U
86 embryo_86 U
87 embryo_87 G
88 embryo_88 G
89 embryo_89 G
90 embryo_90 G
91 embryo_91 G
92 embryo_92 U
93 embryo_93 U
94 embryo_94 U
95 embryo_95 G
96 embryo_96 G
97 embryo_97 U
98 embryo_98 U
99 embryo_99 G
100 embryo_100 U
101 embryo_101 G
102 embryo_102 G
103 embryo_103 U
104 embryo_104 U
105 embryo_105 U
106 embryo_106 G
107 embryo_107 U
108 embryo_108 U
109 embryo_109 U
110 embryo_110 U
111 embryo_111 U
112 embryo_112 G
113 embryo_113 G
114 embryo_114 U
115 embryo_115 U
116 embryo_116 G
117 embryo_117 G
118 embryo_118 U
119 embryo_119 G
120 embryo_120 G
121 embryo_121 U
122 embryo_122 G
123 embryo_123 G
124 embryo_124 U
125 embryo_125 U
126 embryo_126 U
127 embryo_127 G
128 embryo_128 U
# Valor de p no experimento originalp <-0.057# número de embriões em cada grupodf <-40# O teste é bicaudal porque as autoras apenas medem se há diferença, não se é maior ou menor o controle.t <-qt(p/2, df, lower.tail =FALSE) # teste bicaudal por isso o p / 2 (direito e esquerdo)t
[1] 1.959903
# Estimando o tamanho de efeito a a partir dos dados do artigo.# Usando a funcão t_to_d do pacote effectsize podemos calcular o tamamho de efeito # e o intervalo de confiança.effectsize::t_to_d(t, df = df)
d | 95% CI
--------------------
0.62 | [-0.02, 1.25]
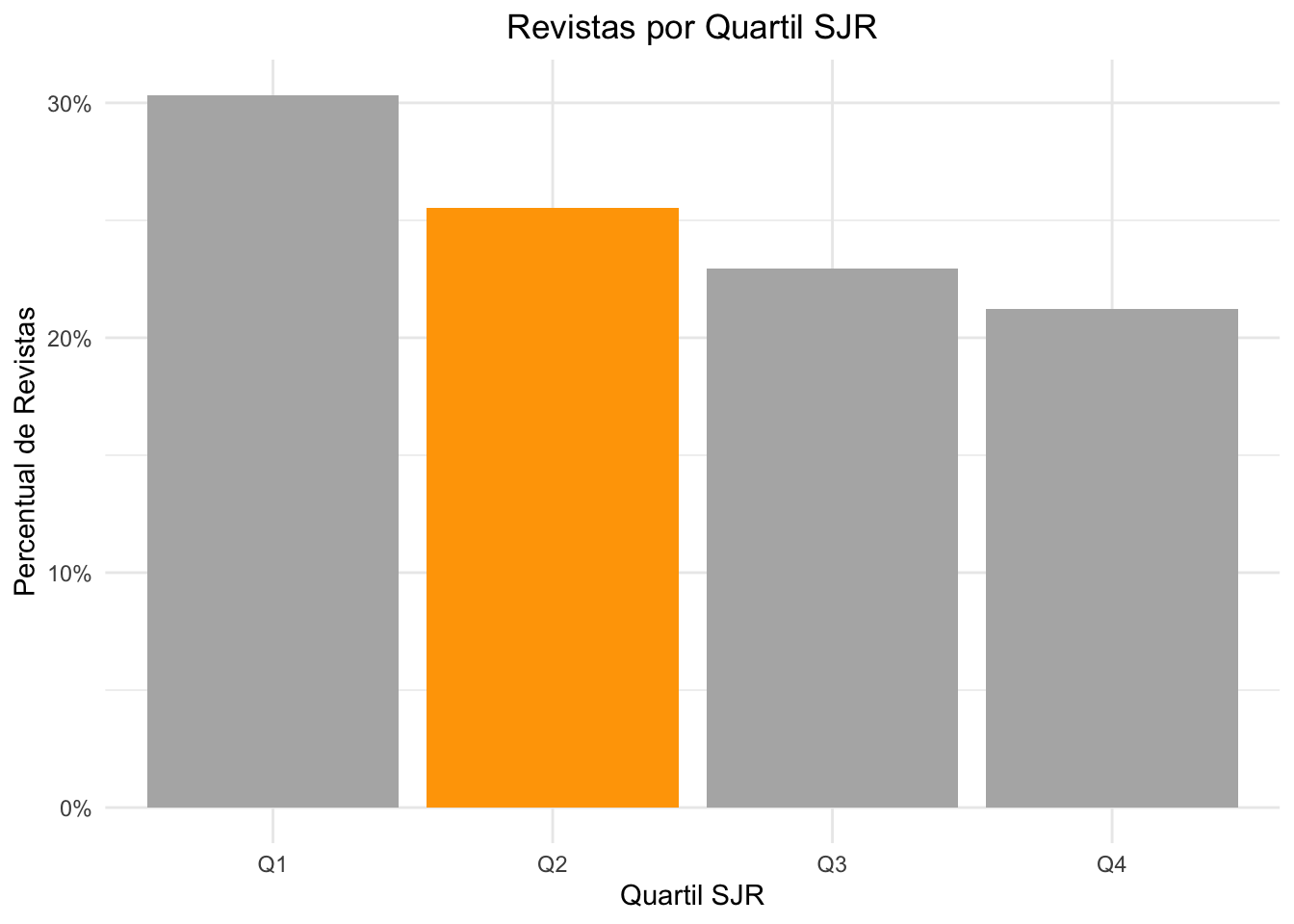
# Vamos criar um "data.frame" a partir de um filtro de busca no dataset.# Os filtros serão por todas as revistas que apresentam classificação, ou seja# a coluna, agora renomeada, best_quartile, não tenha NA e não seja igual a '-'# contabilizamos por best_quartile e para mostrar em valores percentuais# aplicamos uma operação de mutate para substituir o valor do count por# frequencia relativa (n / sum(n)) de cada quartil.quart_counts <- df_sjr |>filter(!is.na(best_quartile), best_quartile !="-") |>count(best_quartile) |>mutate(freq = n /sum(n))# Usamos o ggplot para criar o gráfico de colunas e aplicamos configurações# no gráfico para colocar todas as colunas em cinza, mas a coluna Q2, que é# a coluna de interesse ficar na cor laranja. Poderiamos usar qualquer cor# inclusive em códigos RGB, mas para simplificar, vamos usar nomes das cores no R# e temos: Q2 em laranja e todas as demais em cinza a 70%# No ggplot2, usamos a função aes (de aesthetics) para formatar o gráfico. Com aesthetics,# iremos de fato 'formatar' o gráfico. Por exemplo: do dataset, qual coluna usaremos para# o eixo x, e qual o eixo y. E assim temos: x = best_quartile (se não tivesse renomeado, seria# só com "escapes" para usar o nome), em y usaremos a coluna freq (criada na função mutate).# E dizemos mais: fill (preencher), iremos preencher apenas a coluna best_quartile onde o label for Q2# Mas para que as colunas fiquem com uma cor, usaremos o modificador scale_fill_manual() com apenas# duas cores, pois a Q2 o ggplot irá colorir, as demais serão na cor "nao selecionada" no caso gray70ggplot(quart_counts, aes(x = best_quartile, y = freq, fill = best_quartile =="Q2")) +geom_col() +# gráfico de barras (col=column)scale_y_continuous(labels = scales::percent_format()) +# vamos formar o eixy Y para valores em percentuaisscale_fill_manual(values =c("grey70", "orange")) +# aqui dizemos qual cor para as colunaslabs(title ="Revistas por Quartil SJR",x ="Quartil SJR",y ="Percentual de Revistas" ) +theme_minimal() +# auto explicativotheme(plot.title =element_text(hjust =0.5)) +#aqui ajustamos o titulo para ficar centralizadotheme(legend.position ="none") # não queremos legenda no gráfico